ancient_trichuris
DNA Damage
Author: Stephen Doyle, stephen.doyle[at]sanger.ac.uk
Contents
- pmdtools
- plot deamination frequencies
-
trim bases from reads in bam to remove deamination
- need to check to what degree deanimation (increased frequency of C > T and G > A) has affected the DNA used for sequencing
- this is a common artefact in ancient samples - this is expected from ancient reads, and will likely affect the older samples more.
- Not expecting this in the modern samples
pmdtools
# To view deamination-derived damage patterns in a simple table, without separating CpG sites
#samtools view AN_DNK_COG_EN_002.bam | python pmdtools.0.60.py --deamination
mkdir ${WORKING_DIR}/03_MAPPING/DEAMINATION
# modern samples
while read -r OLD_NAME NEW_NAME; do
# To compute deamination-derived damage patterns separating CpG and non-CpG sites
samtools view ${NEW_NAME}.bam | pmdtools --platypus --requirebaseq 30 --number=10000 > PMD_temp.txt ;
# make plots
R CMD BATCH ${WORKING_DIR}/00_SCRIPTS/plotPMD.R ;
# move and rename
mv deamination_plot.png ${WORKING_DIR}/03_MAPPING/DEAMINATION/${NEW_NAME}.deamination_plot.png ;
done < ${WORKING_DIR}/modern.sample_list
# ancient samples
while read -r NEW_NAME; do
# To compute deamination-derived damage patterns separating CpG and non-CpG sites
samtools view ${NEW_NAME}.bam | pmdtools --platypus --requirebaseq 30 --number=10000 > PMD_temp.txt ;
# make plots
R CMD BATCH ${WORKING_DIR}/00_SCRIPTS/plotPMD.R ;
# move and rename
mv deamination_plot.png ${WORKING_DIR}/03_MAPPING/DEAMINATION/${NEW_NAME}.deamination_plot.png ;
done < ../ancient.sample_list_v2
plot deamination frequencies
- where “plotPMD.R” is:
# script to make deamination frequency plots from PMD data
# load libraries
require(reshape2)
require(patchwork)
require(ggplot2)
require(dplyr)
require(tidyverse)
# read data from PMD, example "samtools view AN_DNK_COG_EN_002.bam | head -n 100000 | pmdtools --platypus --requirebaseq 30 > PMD_temp.txt"
data <- read.table("PMD_temp.txt", header=T)
data2 <- melt(data,id.vars="z")
# split into 5' and 3' datasets, and add some variables for plotting
data_5 <- data2 %>%
filter(str_detect(variable, "5")) %>%
mutate(base_substitution = if_else(str_detect(variable,"GA"), "G-to-A", if_else(str_detect(variable,"CT"), "C-to-T", "other"))) %>%
mutate(CpG_state = if_else(str_detect(variable,"CpG"), "CpG", "non-CpG"))
data_3 <- data2 %>%
filter(str_detect(variable, "3")) %>%
mutate(base_substitution = if_else(str_detect(variable,"GA"), "G-to-A", if_else(str_detect(variable,"CT"), "C-to-T", "other"))) %>%
mutate(CpG_state = if_else(str_detect(variable,"CpG"), "CpG", "non-CpG"))
# make plots
plot_5 <- ggplot(data_5, aes(z, value, colour = base_substitution, group = variable, linetype = CpG_state, size = base_substitution)) +
geom_line() +
ylim(0, 0.2) +
theme_bw() +
scale_colour_manual(values = c("other" = "black", "C-to-T" = "red" , "G-to-A" = "blue"))+
scale_size_manual(values=c("other" = 0.5, "C-to-T" = 1 , "G-to-A" = 1))+
scale_linetype_manual(values = c("CpG" = "dashed", "non-CpG" = "solid")) +
labs(x="Distance from 5' end of sequence read", y="Mismatch frequency")
plot_3 <- ggplot(data_3, aes(z, value, colour = base_substitution, group = variable, linetype = CpG_state, size = base_substitution)) +
geom_line() +
ylim(0, 0.2) +
theme_bw() +
scale_colour_manual(values = c("other" = "black", "C-to-T" = "red" , "G-to-A" = "blue"))+
scale_size_manual(values=c("other" = 0.5, "C-to-T" = 1 , "G-to-A" = 1))+
scale_linetype_manual(values = c("CpG" = "dashed", "non-CpG" = "solid")) +
labs(x="Distance from 3' end of sequence read", y="Mismatch frequency")
# bring it together
plot_5 + plot_3 + plot_layout(guides = "collect")
# save it
ggsave("deamination_plot.png", height=5, width=10)
- Example of a modern sample (MN_CHN_GUA_HS_001)
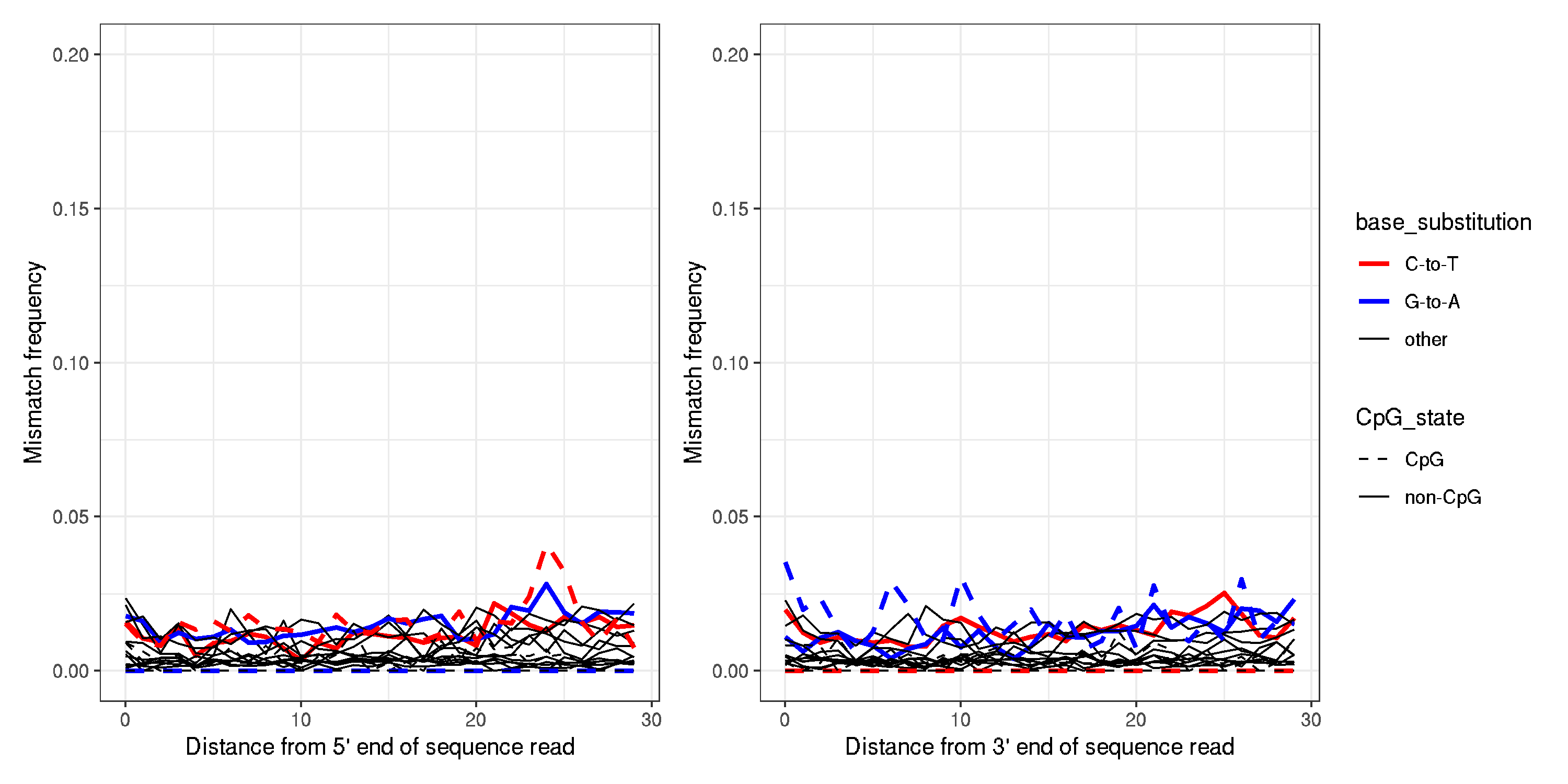
-
Example of a ancient sample (AN_DNK_COG_EN_002)
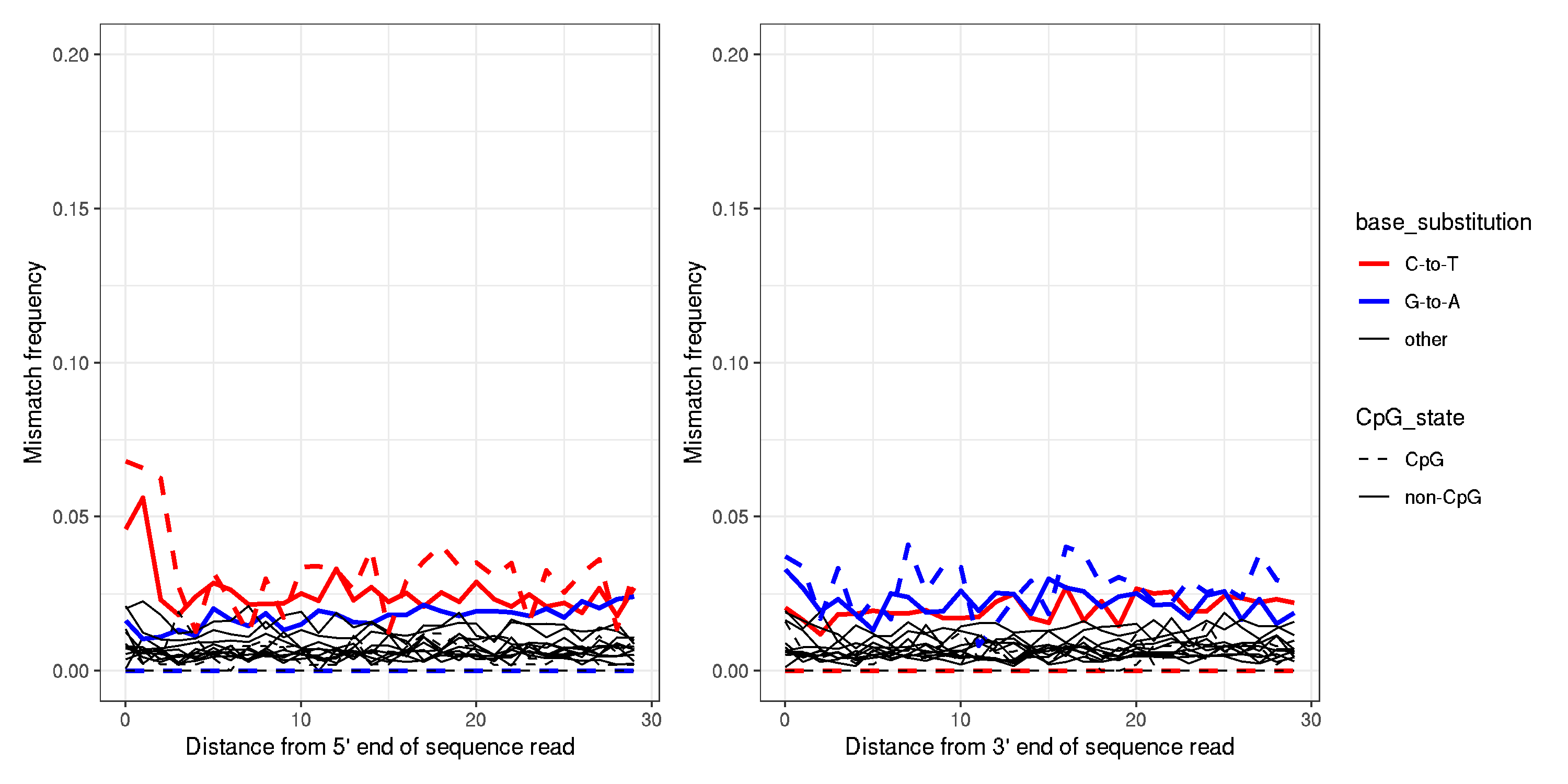
- clearly a CT bias in the first two bases of the ancient sample, that doesnt seem to be present in the modern sample
- not present in all samples I dont think, but in quite a few that I have checked.
- simplest solution is to remove the first two bases from all reads before moving forward
# can calculate the proportion of deamination damage in the 1st position as a proxy for overall damage to the reads
ls -1 *bam | grep -v "trimmed" | while read -r BAM ; do
data=$(samtools view ${BAM} | pmdtools --first --requirebaseq 30 --number=10000); echo -e "${BAM}\t${data}";
done
trim bases from reads in bam
Using “bamUtils trimBam” to remove the 5’ and 3’ 2 bp from mapped reads
# trim modern samples
while read -r OLD_NAME NEW_NAME; do
bsub.py 5 --threads 4 trim_bams "${WORKING_DIR}/00_SCRIPTS/run_trimreads_in_bam.sh ${NEW_NAME}";
done < ${WORKING_DIR}/modern.sample_list
# trim ancient samples
while read -r NEW_NAME; do
bsub.py 5 --threads 4 trim_bams "${WORKING_DIR}/00_SCRIPTS/run_trimreads_in_bam.sh ${NEW_NAME}";
done < ${WORKING_DIR}/ancient.sample_list_v2
# once done, clean up
rm *[0-9].bam*
where “run_trimreads_in_bam.sh” is:
#!/bin/bash
# trim left and right bases from reads in a bam.
NAME=${1}
bamutils_clip_left=2
bamutils_clip_right=2
bamUtils trimBam ${NAME}.bam ${NAME}.tmp.bam -L ${bamutils_clip_left} -R ${bamutils_clip_right}
samtools sort -@ 4 ${NAME}.tmp.bam -o ${NAME}.trimmed.bam
samtools index ${NAME}.trimmed.bam
rm ${NAME}.tmp.bam