ancient_trichuris
Genome coverage
Author: Stephen Doyle, stephen.doyle[at]sanger.ac.uk
Contents
- Genome wide coverage
- Generate quantitative stats on coverage for supplementary tables
- generate some coverage plots
- Genome-wide coverage to determine worm sex
Genome wide coverage
cd /nfs/users/nfs_s/sd21/lustre118_link/trichuris_trichiura/03_MAPPING/COV_STATS
# run coverage stats
bsub.py --queue long 5 cov "~sd21/bash_scripts/run_cov_stats 100000"
where “run_cov_stats” is:
##########################################################################################
# run_cov_stats
##########################################################################################
# Usage: ~sd21/bash_scripts/run_cov_stats < window size >
module load bamtools/2.5.1--he860b03_5
WINDOW=$1
for i in *.bam
do
bamtools header -in ${i} | grep "^@SQ" | awk -F'[:\t]' '{printf $3"\t"1"\t"$5"\n"}' OFS="\t" > ${i%.bam}.chr.bed
bamtools header -in ${i} | grep "^@SQ" | awk -F'[:\t]' '{printf $3"\t"$5"\n"}' OFS="\t" > ${i%.bam}.chr.genome
bedtools makewindows -g ${i%.bam}.chr.genome -w ${WINDOW} > ${i%.bam}.${WINDOW}_window.bed
samtools bedcov -Q 20 ${i%.bam}.chr.bed ${i} | awk -F'\t' '{printf $1"\t"$2"\t"$3"\t"$4"\t"$4/($3-$2)"\n"}' OFS="\t" > ${i%.bam}.chr.cov
samtools bedcov -Q 20 ${i%.bam}.${WINDOW}_window.bed ${i} | awk -F'\t' '{printf $1"\t"$2"\t"$3"\t"$4"\t"$4/($3-$2)"\n"}' OFS="\t" > ${i%.bam}.${WINDOW}_window.cov
rm ${i%.bam}.chr.bed ${i%.bam}.${WINDOW}_window.bed ${i%.bam}.chr.genome
done
for i in *.chr.cov; do
printf "${i}\n" > ${i}.tmp | awk '{print $5}' OFS="\t" ${i} >> ${i}.tmp;
done
paste *.tmp > coverage_stats.summary
rm *.tmp
Generate quantitative stats on coverage for supplementary tables etc
# extract mtDNA and nuclear (mean & stddev) data
for i in *trimmed.chr.cov; do
name=${i%.trimmed.chr.cov};
nuc=$(grep -v "MITO" ${i%.trimmed.chr.cov}.100000_window.cov | datamash mean 5 sstdev 5 );
mtDNA=$(grep "MITO" ${i} | cut -f5 );
echo -e "${name}\t${nuc}\t${mtDNA}";
done > nuc_mtDNA_coverage.stat
- this data will go into a supplementary table
generate some coverage plots,
- particularly to compare relative coverage between sex chromosome scaffolds and autosomes to determine sex of individual sample
# working dir: /nfs/users/nfs_s/sd21/lustre118_link/trichuris_trichiura/03_MAPPING/COV_STATS
# load libraries
library(tidyverse)
# function to plot coverage per individual sample
plot_cov <- function(data, title){
data <- read.table(data,header=F)
plot <- ggplot(data,aes(1:nrow(data),V5,col=V1)) +
geom_point() +
labs(title=title, x="Genomic position", y="Mean coverage per 100kb window") +
theme_bw() + theme(legend.position = "none") +
ylim(0,2*mean(data$V5))
print(plot)
}
# run function, reading in a specific sample, to plot coverage
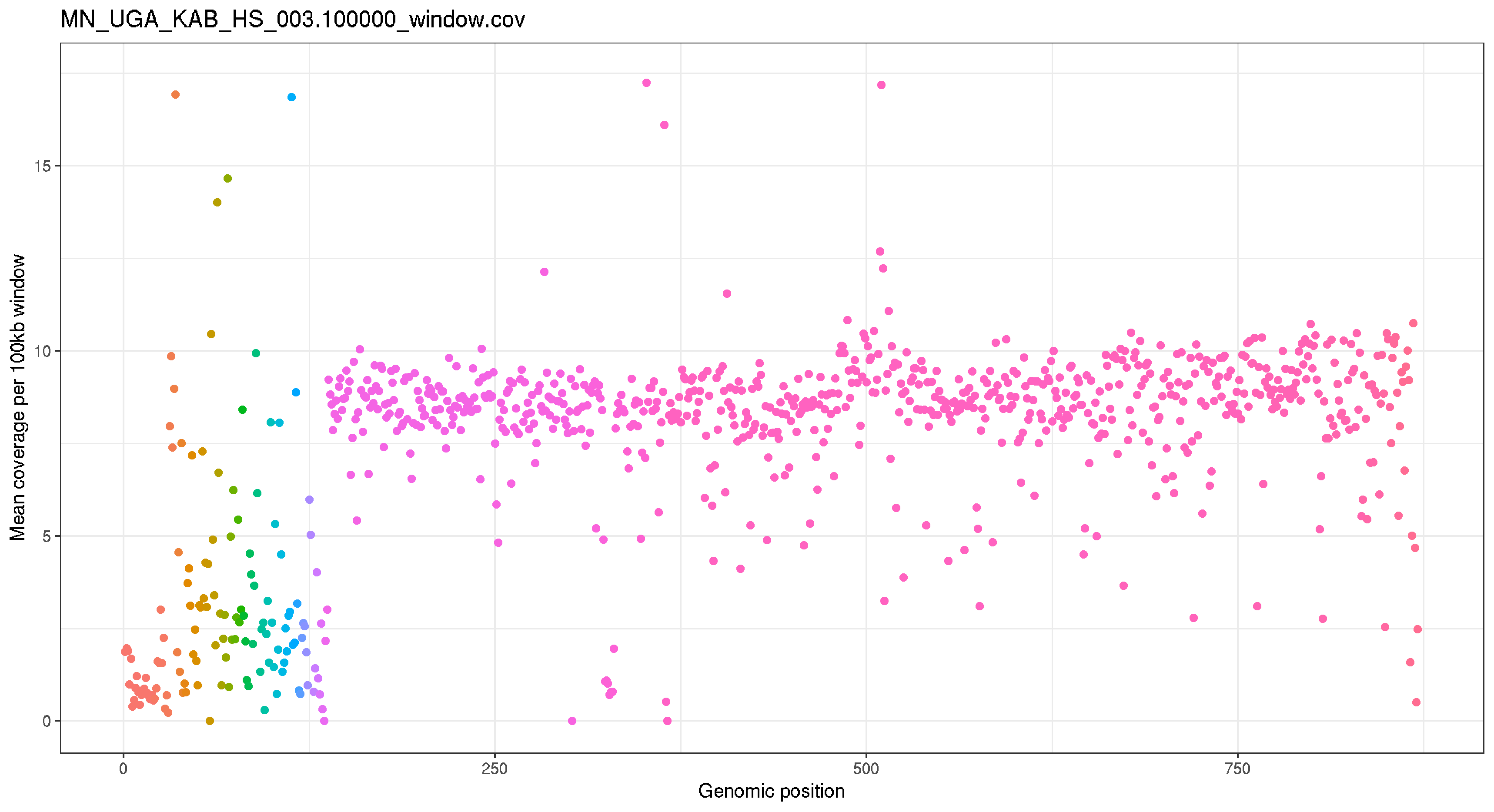
plot_cov("MN_UGA_KAB_HS_003.100000_window.cov","MN_UGA_KAB_HS_003.100000_window.cov")
ggsave("MN_UGA_KAB_HS_003.100000_window.cov.png")
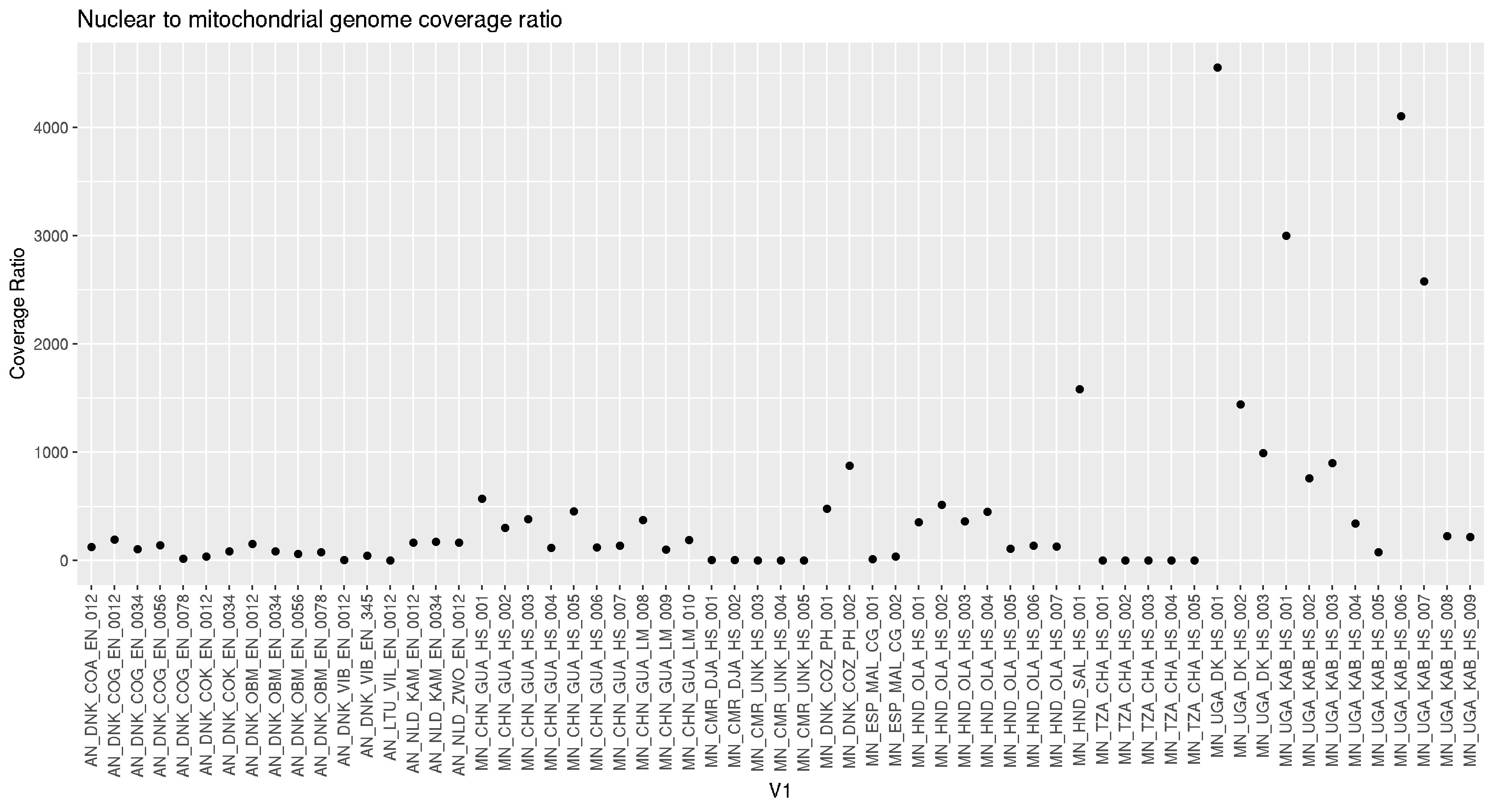
# nuclear to mitochondrial DNA coverage ratio
nucmito <- read.table("nuc_mtDNA_coverage.stats",header=F)
ggplot(nucmito,aes(V1,V4)) +
geom_point() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1)) +
labs(title = "Nuclear to mitochondrial genome coverage ratio", y = "Coverage Ratio")
ggsave("nuc_mito_cov_ratio.png")
Genome-wide coverage to determine worm sex
- using genome wide coverage data to iner the sex of the worms.
- we know that “Trichuris_trichiura_1” lingage group is the X chromosome based on synteny with Trichuris muris
- a ratio of this LG to other scaffolds should give the sex of the individual worms,
- the pooled worms will be mixed sex, so should have an intermediate profile to that of either male or female individual worms.
### Make some genome wide coverage plots
# load libraries
library(tidyverse)
library(ggsci)
library(stringr)
# list file names
file_names <- list.files(path = "./",pattern = ".trimmed.100000_window.cov")
# load data using file names, and make a formatted data frame
data <- purrr::map_df(file_names, function(x) {
data <- read.delim(x, header = F, sep="\t")
data <- tibble::rowid_to_column(data, "NUM")
cbind(sample_name = gsub(".trimmed.100000_window.cov","",x), data)
})
colnames(data) <- c("sample_name", "NUM", "CHROM", "START", "END", "RAW_COVERAGE", "PROPORTION_COVERAGE")
# remove mitochondrial genome and unplaced scaffolds
data <- data[data$CHROM != "Trichuris_trichiura_MITO",]
data <- data %>% filter(!str_detect(CHROM, "Trichuris_trichiura_00_"))
# annotate sex linked scaffolds
data$SEX <- str_detect(data$CHROM,"Trichuris_trichiura_1_")
# plot boxplots and distributions of pairwise Fst analyses
ggplot(data, aes(NUM, PROPORTION_COVERAGE/(median(PROPORTION_COVERAGE)), col=SEX, group = sample_name)) +
geom_point(size=0.2) +
labs( x = "Genome position" , y = "Relative coverage per 100kb window") +
theme_bw() + theme(legend.position = "none", strip.text.x = element_text(size = 6)) +
facet_wrap(~sample_name, scales = "free_y")+scale_color_manual(values=rep(c("orange","blue"),2))+ theme_classic()
ggsave("plot_relative_genomewide_coverage_allsamples.png")
ggsave("plot_relative_genomewide_coverage_allsamples.pdf",height=10, width=20, useDingbats=FALSE)
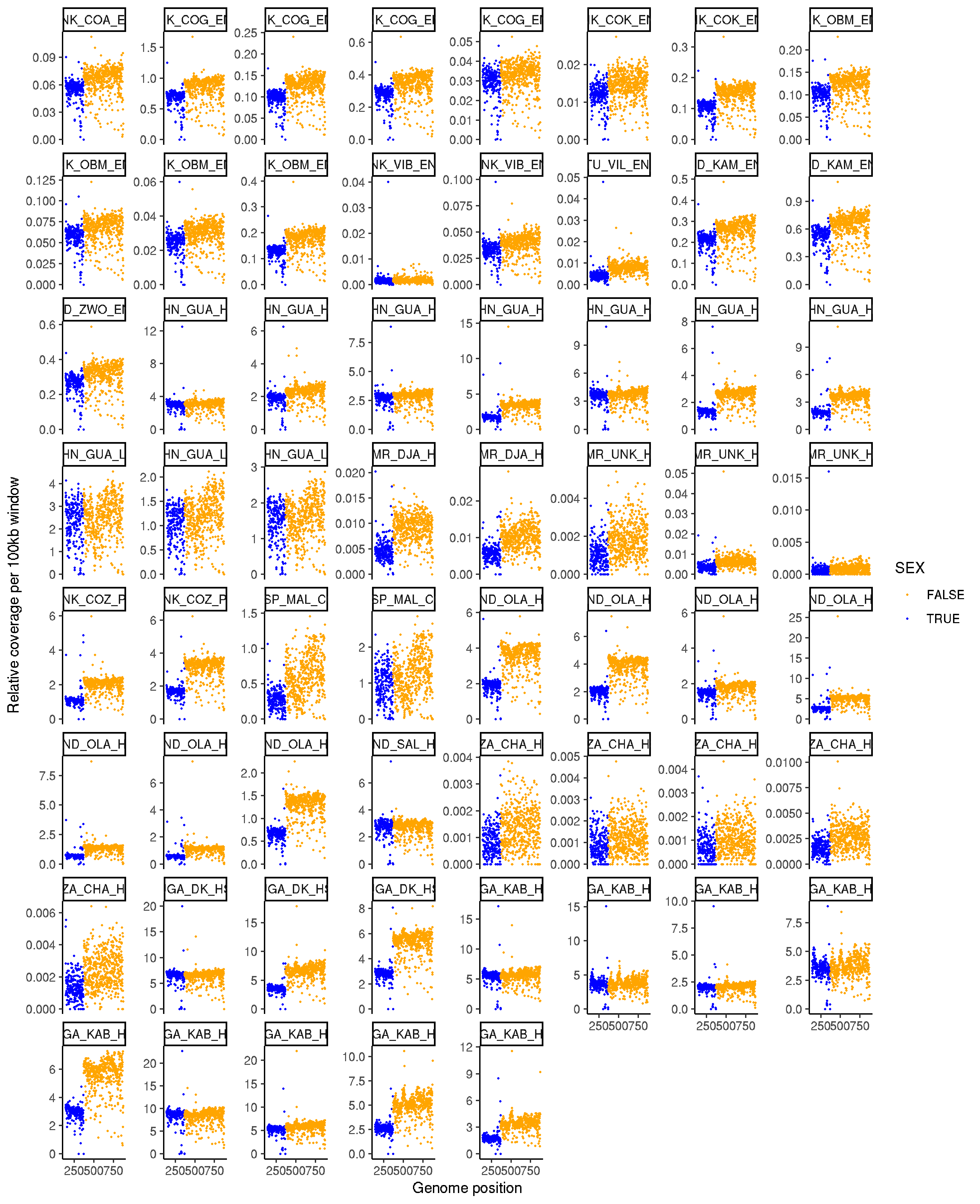
- originally made these plots to determine relative coverage of X to autosomes, but figured it’d be better simply to calculate the ratio and plot it, so I have dont that in the next section.
# extract mtDNA and nuclear (mean & stddev) data
for i in *trimmed.chr.cov; do
name=${i%.trimmed.chr.cov};
autosome=$(grep "Trichuris_trichiura_2\|Trichuris_trichiura_3]" ${i%.trimmed.chr.cov}.100000_window.cov | datamash mean 5 sstdev 5 );
xchromosome=$(grep "Trichuris_trichiura_1" ${i%.trimmed.chr.cov}.100000_window.cov | datamash mean 5 sstdev 5 );
echo -e "${name}\t${autosome}\t${xchromosome}";
done > autosome_to_Xchromsome_cov.stat
- and to plot the data
library(tidyverse)
cov_data <- read.table("autosome_to_Xchromsome_cov.stat")
sample_type <- read.table("single_v_pooled_sampletype.txt")
data <- left_join(cov_data,sample_type,by="V1")
colnames(data) <- c("sample_name", "autosome_cov_mean", "autosome_cov_sd", "x_cov_mean", "x_cov_sd", "sample_type" )
ggplot(data, aes(sample_name, x_cov_mean/autosome_cov_mean, col=sample_type)) +
geom_point() +
labs(title="X-to-autosomal coverage ratio", x="Sample name", y="X-to-autosomal coverage ratio") +
theme_bw() + theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1)) +
ylim(0.35,1.2) +
coord_flip()
ggsave("plot_x-to-autosome_ratio_sexdet.png")
ggsave("plot_x-to-autosome_ratio_sexdet.pdf",height=10, width=7, useDingbats=FALSE)
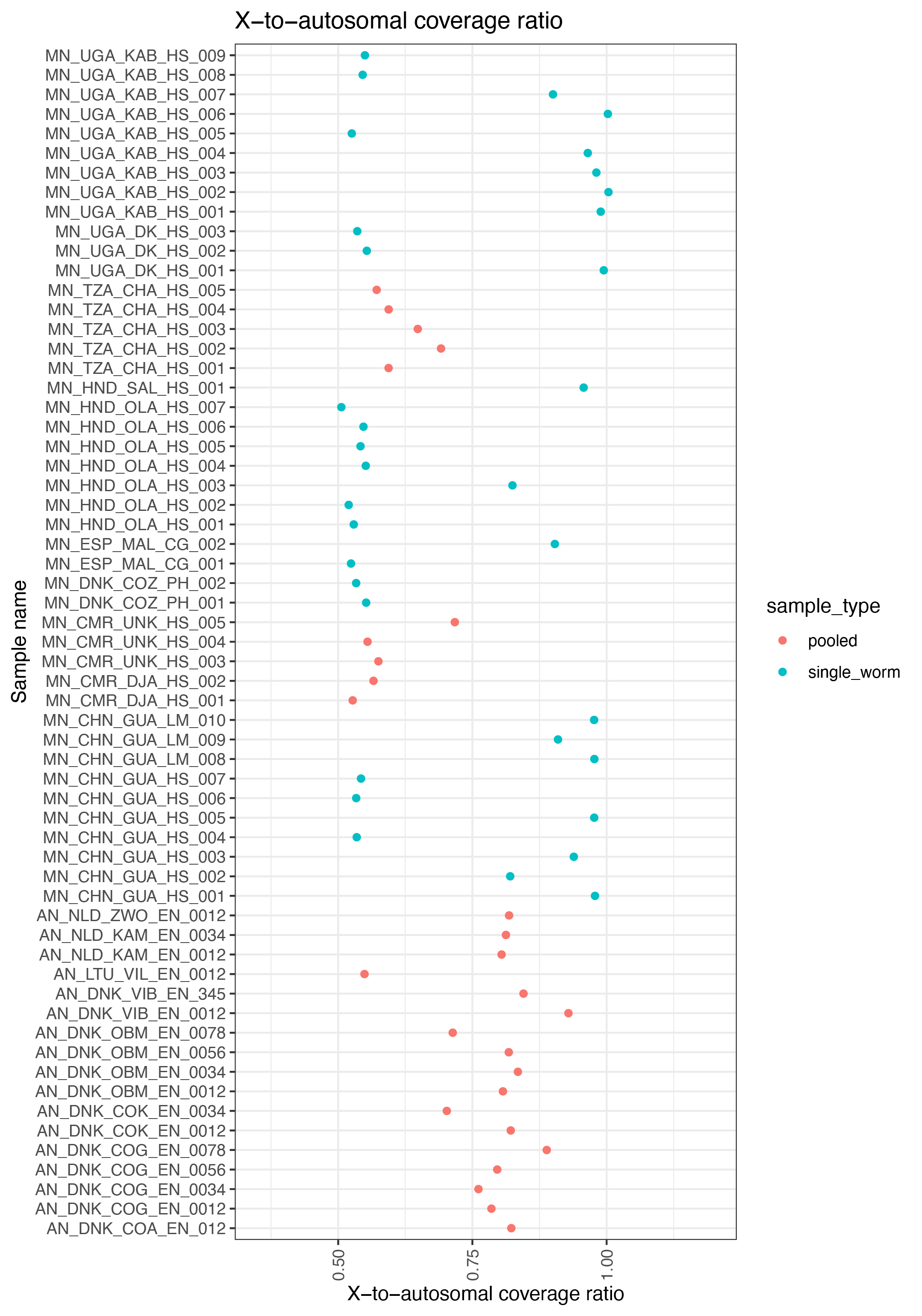
- count 17 males based on ~0.5x coverage (XY)
- count 17 females based on ~1X coverage
- note 2 with intermediate coverage, likely female but not clear