ancient_trichuris
Principle component analysis to explore genetic diversity
Author: Stephen Doyle, stephen.doyle[at]sanger.ac.uk
Contents
- PCA of mitochondrial variants
- PCA of nuclear variants
PCA of mitochondrial variation
# working directory
/nfs/users/nfs_s/sd21/lustre118_link/trichuris_trichiura/05_ANALYSIS/PCA
# load libraries
library(tidyverse)
library(gdsfmt)
library(SNPRelate)
library(ggsci)
snpgdsClose(genofile)
vcf.in <- "mito_samples3x_missing0.8.recode.vcf.gz"
gds <- snpgdsVCF2GDS(vcf.in, "mtDNA_1.gds", method="biallelic.only")
genofile <- snpgdsOpen(gds)
pca <- snpgdsPCA(genofile, num.thread=2, autosome.only = F)
samples <- as.data.frame(pca$sample.id)
colnames(samples) <- "name"
metadata <- samples %>% separate(name,c("time", "country","population","host","sampleID"))
metadata <- metadata %>%
mutate(SAMPLE_AGE = ifelse(time == "AN", "Ancient",
ifelse(host == "HS", "Modern Human", "Modern Animal")))
data <-
data.frame(sample.id = pca$sample.id,
EV1 = pca$eigenvect[,1],
EV2 = pca$eigenvect[,2],
EV3 = pca$eigenvect[,3],
EV4 = pca$eigenvect[,4],
EV5 = pca$eigenvect[,5],
EV6 = pca$eigenvect[,6],
TIME = metadata$SAMPLE_AGE,
COUNTRY = metadata$country,
POPULATION = metadata$population,
HOST = metadata$host,
stringsAsFactors = FALSE)
country_colours <-
c("CHN" = "#00A087",
"CMR" = "#902F21",
"DNK" = "#3C5488",
"ESP" = "#E7EAF0",
"HND" = "#4DBBD5",
"NLD" = "#9DAAC4",
"UGA" = "#E64B35",
"LTU" = "#0F1522",
"TZA" = "#F2A59A")
plot_pca <-
ggplot(data, aes(EV1, EV2, col = COUNTRY, shape = TIME, label = COUNTRY)) +
geom_text(size=4) +
theme_bw() +
labs(title="mito_samples3x_missing0.8",
x = paste0("PC1 variance: ",round(pca$varprop[1]*100,digits=2),"%"),
y = paste0("PC2 variance: ",round(pca$varprop[2]*100,digits=2),"%"))+
scale_colour_manual(values = country_colours)
plot_pca
ggsave("plot_PCA_mito_samples3x_missing0.8.png")
# use this to label with sample names
#ggplot(tab,aes(EV1,EV2, col = COUNTRY, shape = TIME, label = COUNTRY)) + geom_point(size=4)
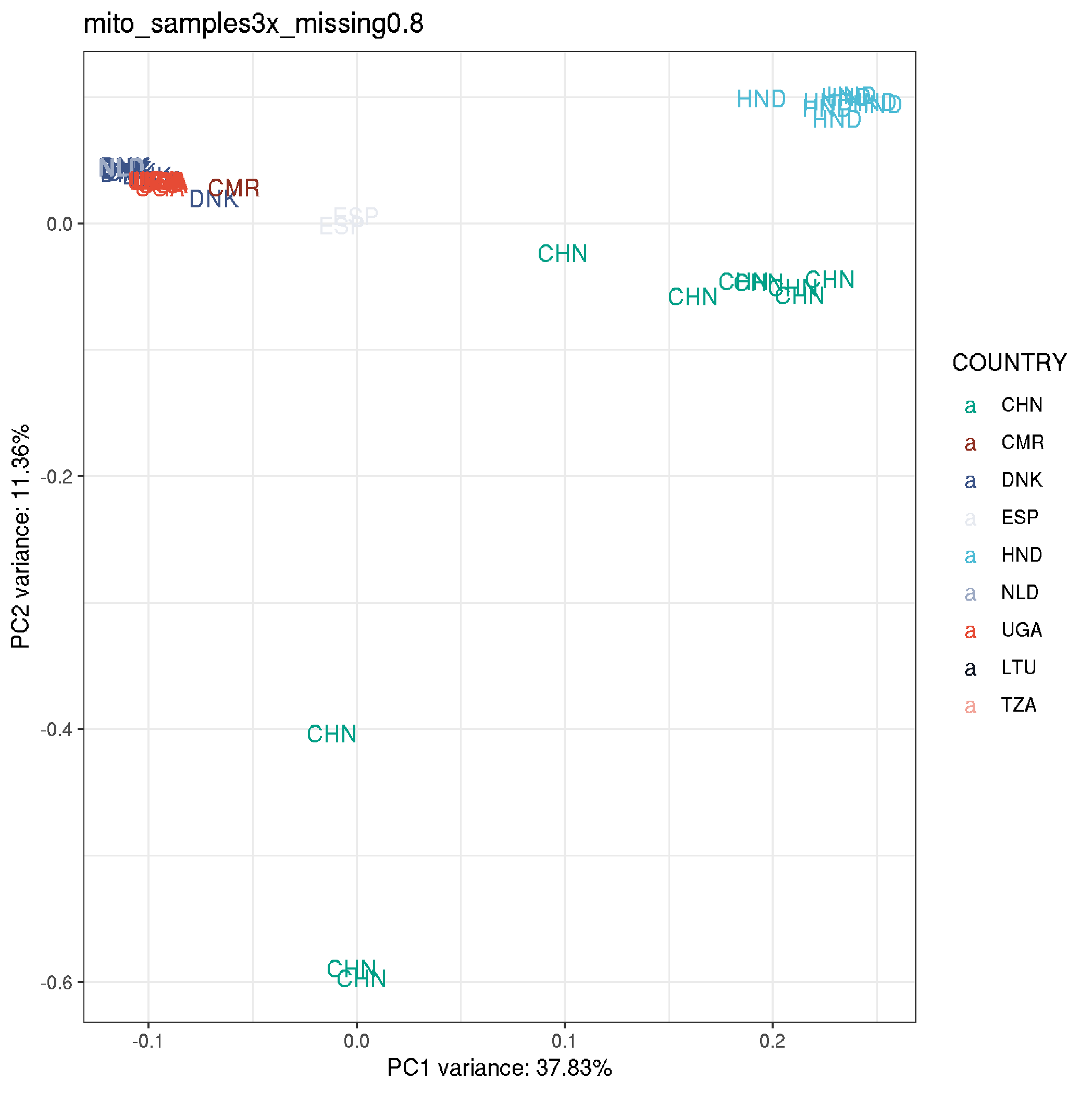
- the CHN LF samples are the outliers in the above plot, so rerunning to remove these
vcftools \
--gzvcf Trichuris_trichiura.cohort.mito_variants.final.recode.vcf.gz \
--keep mtDNA_3x_noLF.list \
--max-missing 0.8 \
--recode --recode-INFO-all \
--out mito_samples3x_missing0.8_noLF
gzip -f mito_samples3x_missing0.8_noLF*
#> After filtering, kept 48 out of 61 Individuals
#> After filtering, kept 1291 out of a possible 1888 Sites
# load libraries
library(tidyverse)
library(gdsfmt)
library(SNPRelate)
library(ggsci)
library(patchwork)
snpgdsClose(genofile)
vcf.in <- "mito_samples3x_missing0.8_noLF.recode.vcf.gz"
gds <- snpgdsVCF2GDS(vcf.in, "mtDNA.gds", method="biallelic.only")
genofile <- snpgdsOpen(gds)
pca <- snpgdsPCA(genofile, num.thread=2, autosome.only = F)
# Working space: 56 samples, 802 SNPs
samples <- as.data.frame(pca$sample.id)
colnames(samples) <- "name"
metadata <- samples %>% separate(name,c("time", "country","population","host","sampleID"))
metadata <- metadata %>%
mutate(SAMPLE_AGE = ifelse(time == "AN", "Ancient",
ifelse(host == "HS", "Modern Human", "Modern Animal")))
data <-
data.frame(sample.id = pca$sample.id,
EV1 = pca$eigenvect[,1],
EV2 = pca$eigenvect[,2],
EV3 = pca$eigenvect[,3],
EV4 = pca$eigenvect[,4],
EV5 = pca$eigenvect[,5],
EV6 = pca$eigenvect[,6],
TIME = metadata$SAMPLE_AGE,
COUNTRY = metadata$country,
POPULATION = metadata$population,
HOST = metadata$host,
stringsAsFactors = FALSE)
country_colours <-
c("CHN" = "#00A087",
"CMR" = "#902F21",
"DNK" = "#3C5488",
"ESP" = "#E7EAF0",
"HND" = "#4DBBD5",
"NLD" = "#9DAAC4",
"UGA" = "#E64B35",
"LTU" = "#0F1522",
"TZA" = "#F2A59A")
plot_pca_mito <-
ggplot(data, aes(EV1, EV2, col = COUNTRY, shape = TIME, label = COUNTRY),alpha=1) +
geom_rect(aes(xmin=-0.125, ymin=-0.01, xmax=-0.05, ymax=0.025), fill=NA, col="black", linetype="dotted", size=0.3) +
geom_point(size=4, alpha=1) +
theme_bw() +
labs(x = paste0("PC1 variance: ",round(pca$varprop[1]*100,digits=2),"%"),
y = paste0("PC2 variance: ",round(pca$varprop[2]*100,digits=2),"%")) +
scale_colour_manual(values = country_colours)
plot_pca_mito_zoom <-
ggplot(data, aes(EV1,EV2, col = COUNTRY, shape = TIME, label = paste0(TIME,"_",COUNTRY,"_",POPULATION,"_",HOST))) +
geom_point(size=4, alpha=1) +
theme_bw() +
labs(x = "PC1",
y = "PC2") +
xlim(-0.125,-0.05) + ylim(-0.01, 0.025) +
scale_colour_manual(values = country_colours)
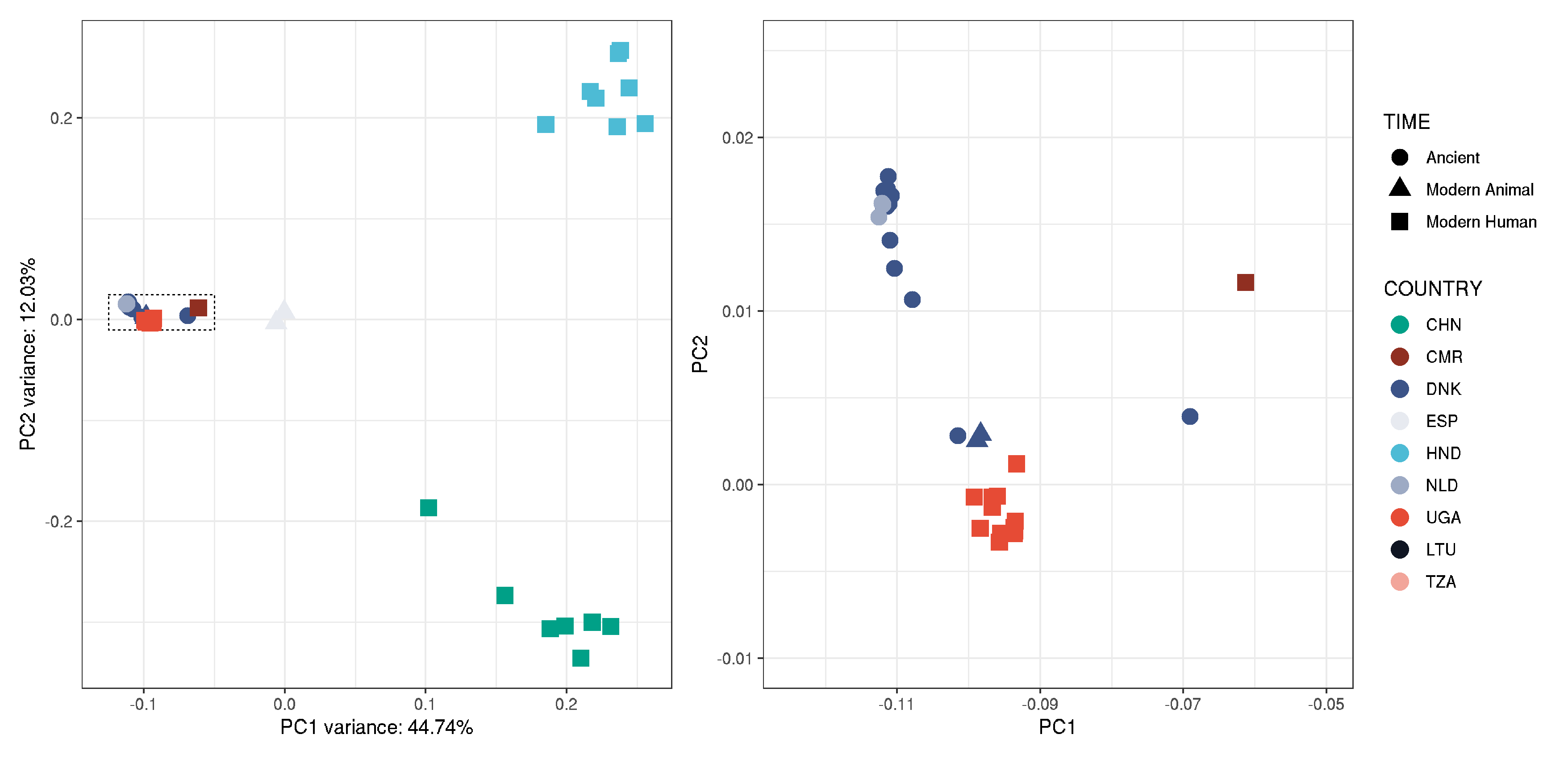
# note: geom_rect in first plot and zoom coordinates were adjusted manually to show the cluster clearly.
plot_pca_mito + plot_pca_mito_zoom + plot_layout(ncol=2, guides = "collect")
ggsave("plot_PCA_mito_samples3x_missing0.8_noLF.png")
ggsave("plot_PCA_mito_samples3x_missing0.8_noLF.pdf", height = 5, width = 11, useDingbats=FALSE)
Figure: plot_PCA_mito_samples3x_missing0.8_noLF
- will uses this in Figure 1 panels A and B
PCA of nuclear variants
- human + animal + 2 ancients
- “nuclear_3x_animal.list”
# extract nuclear variants
# vcftools \
# --gzvcf Trichuris_trichiura.cohort.nuclear_variants.final.recode.vcf.gz \
# --keep nuclear_3x_animal.list \
# --max-missing 0.8 \
# --recode --recode-INFO-all \
# --out nuclear_samples3x_missing0.8
# post peer review - restricted to autosomes only, excluding sex chromosomes
vcftools \
--gzvcf Trichuris_trichiura.cohort.nuclear_variants.final.recode.vcf.gz \
--keep nuclear_3x_animal.list \
--max-missing 0.8 \
--recode --recode-INFO-all \
--bed trichuris_trichiura.autosomeLG.bed \
--out nuclear_samples3x_missing0.8
gzip -f nuclear_samples3x_missing0.8.recode.vcf
#> After filtering, kept 36 out of 61 Individuals
#> After filtering, kept 1850621 out of a possible 6933531 Sites
# load libraries
library(tidyverse)
library(gdsfmt)
library(SNPRelate)
library(patchwork)
snpgdsClose(genofile)
vcf.in <- "nuclear_samples3x_missing0.8.recode.vcf.gz"
gds<-snpgdsVCF2GDS(vcf.in, "nuclear_1.gds", method="biallelic.only")
genofile <- snpgdsOpen(gds)
pca <- snpgdsPCA(genofile, num.thread=2, autosome.only = F)
# Working space: 36 samples, 1,675,392 SNPs
samples <- as.data.frame(pca$sample.id)
colnames(samples) <- "name"
metadata <- samples %>% separate(name,c("time", "country","population","host","sampleID"))
metadata <- metadata %>%
mutate(SAMPLE_AGE = ifelse(time == "AN", "Ancient",
ifelse(host == "HS", "Modern Human", "Modern Animal")))
data <-
data.frame(sample.id = pca$sample.id,
EV1 = pca$eigenvect[,1],
EV2 = pca$eigenvect[,2],
EV3 = pca$eigenvect[,3],
EV4 = pca$eigenvect[,4],
EV5 = pca$eigenvect[,5],
EV6 = pca$eigenvect[,6],
TIME = metadata$SAMPLE_AGE,
COUNTRY = metadata$country,
POPULATION = metadata$population,
HOST = metadata$host,
stringsAsFactors = FALSE)
country_colours <-
c("CHN" = "#00A087",
"CMR" = "#902F21",
"DNK" = "#3C5488",
"ESP" = "#E7EAF0",
"HND" = "#4DBBD5",
"NLD" = "#9DAAC4",
"UGA" = "#E64B35",
"LTU" = "#0F1522",
"TZA" = "#F2A59A")
plot_pca_nuc <-
ggplot(data,aes(EV1, EV2, col = COUNTRY, shape = TIME, label = HOST)) +
geom_point(size=4) +
geom_text_repel(size=4) +
theme_bw() +
labs(title="nuclear_samples3x_missing0.8",
x = paste0("PC1 variance: ",round(pca$varprop[1]*100,digits=2),"%"),
y = paste0("PC2 variance: ",round(pca$varprop[2]*100,digits=2),"%"))+
scale_colour_manual(values = country_colours)
plot_pca_nuc
ggsave("plot_PCA_nuclear_samples3x_missing0.8.png")
ggsave("plot_PCA_nuclear_samples3x_missing0.8.pdf", height=5, width=5)
![]()
- main outliers are colobus and leafmonkey, so will remove and rerun
# vcftools \
# --gzvcf Trichuris_trichiura.cohort.nuclear_variants.final.recode.vcf.gz \
# --keep nuclear_3x_animalPhonly.list \
# --max-missing 0.8 \
# --recode --recode-INFO-all \
# --out nuclear_samples3x_missing0.8_animalPhonly
# post peer review - restricted to autosomes only, excluding sex chromosomes
vcftools \
--gzvcf Trichuris_trichiura.cohort.nuclear_variants.final.recode.vcf.gz \
--keep nuclear_3x_animalPhonly.list \
--max-missing 0.8 \
--recode --recode-INFO-all \
--bed trichuris_trichiura.autosomeLG.bed \
--out nuclear_samples3x_missing0.8_animalPhonly
gzip -f nuclear_samples3x_missing0.8_animalPhonly.recode.vcf
#> After filtering, kept 31 out of 61 Individuals
#> After filtering, kept 1963868 out of a possible 6933531 Sites
# load libraries
library(tidyverse)
library(gdsfmt)
library(SNPRelate)
library(ggsci)
snpgdsClose(genofile)
vcf.in <- "nuclear_samples3x_missing0.8_animalPhonly.recode.vcf.gz"
gds <- snpgdsVCF2GDS(vcf.in, "nuclear.gds", method="biallelic.only")
genofile <- snpgdsOpen(gds)
pca <- snpgdsPCA(genofile, num.thread=2, autosome.only = F)
# Working space: 31 samples, 2,544,110 SNPs
samples <- as.data.frame(pca$sample.id)
colnames(samples) <- "name"
metadata <- samples %>% separate(name,c("time", "country","population","host","sampleID"))
# fix the metadata so can get the right shapes to match the main sampling map
metadata <- metadata %>%
mutate(SAMPLE_AGE = ifelse(time == "AN", "Ancient",
ifelse(host == "HS", "Modern Human", "Modern Animal")))
data <-
data.frame(sample.id = pca$sample.id,
EV1 = pca$eigenvect[,1],
EV2 = pca$eigenvect[,2],
EV3 = pca$eigenvect[,3],
EV4 = pca$eigenvect[,4],
EV5 = pca$eigenvect[,5],
EV6 = pca$eigenvect[,6],
TIME = metadata$SAMPLE_AGE,
COUNTRY = metadata$country,
POPULATION = metadata$population,
HOST = metadata$host,
stringsAsFactors = FALSE)
country_colours <-
c("CHN" = "#00A087",
"CMR" = "#902F21",
"DNK" = "#3C5488",
"ESP" = "#E7EAF0",
"HND" = "#4DBBD5",
"NLD" = "#9DAAC4",
"UGA" = "#E64B35",
"LTU" = "#0F1522",
"TZA" = "#F2A59A")
plot_pca_nuc <-
ggplot(data,aes(EV1, EV2, col = COUNTRY, shape = TIME)) +
geom_point(size=4, alpha=1) +
theme_bw() +
labs(x = paste0("PC1 variance: ",round(pca$varprop[1]*100,digits=2),"%"),
y = paste0("PC2 variance: ",round(pca$varprop[2]*100,digits=2),"%")) +
scale_colour_manual(values = country_colours)
plot_pca_nuc_labels <-
ggplot(data,aes(EV1, EV2, col = COUNTRY, shape = TIME, label = POPULATION)) +
geom_point(size=4, alpha=1) +
geom_text_repel() +
theme_bw() +
labs(x = paste0("PC1 variance: ",round(pca$varprop[1]*100,digits=2),"%"),
y = paste0("PC2 variance: ",round(pca$varprop[2]*100,digits=2),"%")) +
scale_colour_manual(values = country_colours)
plot_pca_nuc
# plot
ggsave("plot_PCA_nuclear_samples3x_missing0.8_animalPhonly.pdf", height = 5, width = 5, useDingbats=FALSE)
ggsave("plot_PCA_nuclear_samples3x_missing0.8_animalPhonly.png")
plot_pca_mito + plot_pca_mito_zoom + plot_pca_nuc + plot_layout(ncol=3, guides = "collect")
ggsave("plot_PCA_mito_mitozoom_nuc.pdf", height = 4, width = 14, useDingbats=FALSE)
ggsave("plot_PCA_plot_PCA_mito_mitozoom_nuc.png")
Figure: plot_PCA_nuclear_samples3x_missing0.8_animalPhonly
- to be used in Figure 1, panel D
![]()
![]()