ancient_trichuris
ANGSD
Author: Stephen Doyle, stephen.doyle[at]sanger.ac.uk
Contents
- IBS and Coviance matrices
- Single allele analyses of mitochondrial and nuclear variants
IBS and Coviance matrices
- exploring the use of ANGSD, whcih can use genotype likelihoods for a number of analyses. Probably good for the low coverage datasets
- started off calculating “identity by state” (IBS), which is another way of showing genetic similarity between samples
cd /nfs/users/nfs_s/sd21/lustre118_link/trichuris_trichiura/05_ANALYSIS/ANGSD
bsub.py --threads 5 10 angsd "/nfs/users/nfs_s/sd21/lustre118_link/software/ANCIENT/angsd/angsd -bam bam.list -minMapQ 30 -minQ 20 -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 2e-6 -doIBS 1 -doCounts 1 -doCov 1 -makeMatrix 1 -minMaf 0.05 -P 5"
# load libraries
library(gplots)
# load data
cov_data <- as.matrix(read.table("angsdput.covMat",header=F))
ibs_data <- as.matrix(read.table("angsdput.ibsMat",header=F))
names <- as.matrix(read.table("sample.names"))
rownames(ibs_data) <- names
colnames(ibs_data) <- names
rownames(cov_data) <- names
colnames(cov_data) <- names
pdf("nuclear_ibs_heatmap.pdf",width=15, height=15)
heatmap.2(ibs_data, trace="none", margins=c(12,12))
dev.off()
pdf("nuclear_covariance_heatmap.pdf",width=15, height=15)
cov_data[cov_data > 1] <- 1
heatmap.2(cov_data, trace="none", margins=c(12,12))
dev.off()
-
IBS for nuclear markers
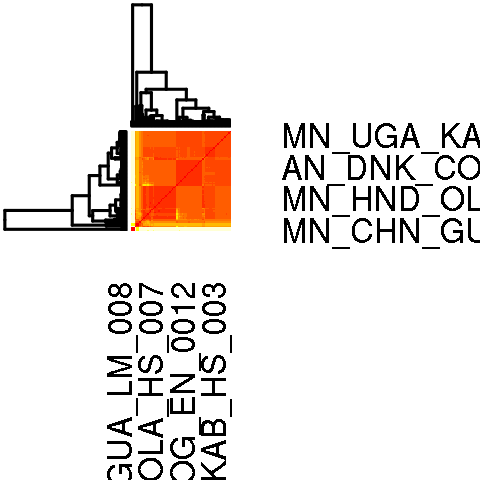
-
covariance of IBS for nuclear markers
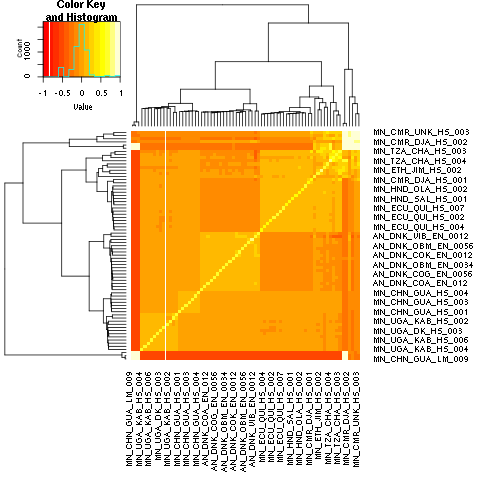
Single allele analyses of mitochondrial and nuclear variants
#mitochondrial variants
zcat ../../04_VARIANTS/GATK_HC_MERGED/mito_samples3x_missing0.8.recode.vcf.gz | cut -f1,2 | grep -v "#" > mito_variable_positions.txt
/nfs/users/nfs_s/sd21/lustre118_link/software/ANCIENT/angsd/angsd sites index mito_variable_positions.txt
bsub.py --queue yesterday 10 mitoANGSD "/nfs/users/nfs_s/sd21/lustre118_link/software/ANCIENT/angsd/angsd -bam mito.bam.list -minMapQ 30 -minQ 20 -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 2e-6 -doIBS 1 -doCounts 1 -doCov 1 -makeMatrix 1 -minMaf 0.05 -P 5 -out mito -sites mito_variable_positions.txt"
# nuclear (autosomal) variants
zcat ../../04_VARIANTS/GATK_HC_MERGED/nuclear_samples3x_missing0.8_animalPhonly.recode.vcf.gz |\
awk '{if($1~/Trichuris_trichiura_2/ || $1~/Trichuris_trichiura_3/) print $1,$2}' OFS="\t" > autosomal_variable_positions.txt
/nfs/users/nfs_s/sd21/lustre118_link/software/ANCIENT/angsd/angsd sites index autosomal_variable_positions.txt
while read NAME; do
ls -1 ../../03_MAPPING/$NAME.trimmed.bam;
done < nuclear_3x_animalPhonly.list > autosomal.bam.list
bsub.py --queue yesterday 10 autoANGSD "/nfs/users/nfs_s/sd21/lustre118_link/software/ANCIENT/angsd/angsd -bam autosomal.bam.list -minMapQ 30 -minQ 20 -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 2e-6 -doIBS 1 -doCounts 1 -doCov 1 -makeMatrix 1 -minMaf 0.05 -P 5 -out autosomal -sites autosomal_variable_positions.txt"
cat mito.bam.list | sed -e 's/.*\///' -e 's/.trimmed.bam//g' | awk -F '[_]' '{print $0,$2,$3}' OFS="\t" > mito.metadata.txt
cat autosomal.bam.list | sed -e 's/.*\///' -e 's/.trimmed.bam//g' | awk -F '[_]' '{print $0,$2,$3}' OFS="\t" > autosomal.metadata.txt
# plots
library(tidyverse)
library(patchwork)
mito_ibs <- read.table("mito.ibsMat")
mito_metadata <- read.table("mito.metadata.txt")
mito_matrix <- as.matrix(mito_ibs)
mito_mds <- cmdscale(as.dist(mito_matrix))
mito_mds <- as.data.frame(mito_mds)
colnames(mito_mds) <- c("mds1","mds2")
mito_data <- cbind(mito_metadata,mito_mds)
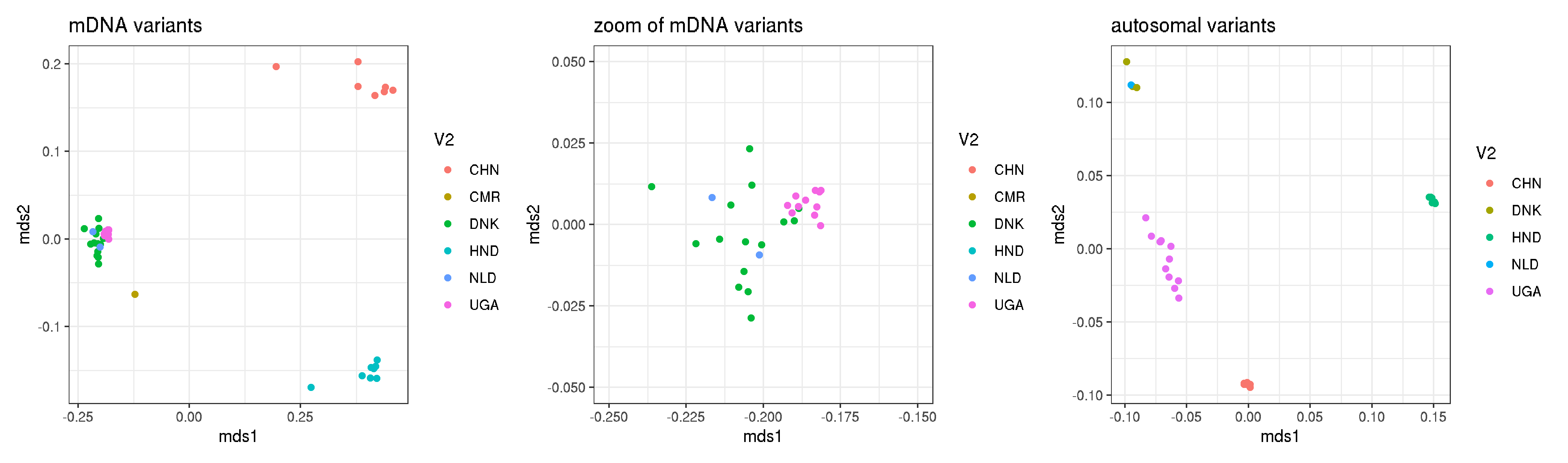
plot_1 <- ggplot(mito_data, aes(mds1,mds2, col=V2)) + geom_point() + theme_bw() + labs(title="mDNA variants")
plot_2 <- ggplot(mito_data, aes(mds1,mds2, col=V2)) + geom_point() + theme_bw() + xlim(-0.25,-0.15) + ylim(-0.05,0.05) + labs(title="zoom of mDNA variants")
autosomal_ibs <- read.table("autosomal.ibsMat")
autosomal_metadata <- read.table("autosomal.metadata.txt")
autosomal_matrix <- as.matrix(autosomal_ibs)
autosomal_mds <- cmdscale(as.dist(autosomal_matrix))
autosomal_mds <- as.data.frame(autosomal_mds)
colnames(autosomal_mds) <- c("mds1","mds2")
autosomal_data <- cbind(autosomal_metadata,autosomal_mds)
plot_3 <- ggplot(autosomal_data, aes(mds1,mds2, col=V2)) + geom_point() + theme_bw() + labs(title="autosomal variants")
plot_1 + plot_2 + plot_3 + plot_layout(ncol=3)
ggsave("plot_mds_single_allele_mito_zoom_nuc.png")
- single allele sampling, using plots consistent with figure 1
- shows that they are almost identical between this plot and figure 1
- conclusion is that the Figure 1 data are not significantly influenced by coverage or missingness