ancient_trichuris
ADMIXTOOLS
Author: Stephen Doyle, stephen.doyle[at]sanger.ac.uk
Contents
- Prepared data and run admixtools
-
plotting admix data
- ADMIXTOOLS is a widely used software package for calculating admixture statistics and testing population admixture hypotheses.
- need to convert vcf to eigenstrat format first
- followed workflow from here: https://speciationgenomics.github.io/ADMIXTOOLS_admixr/
- needed additional scripts
- “convertVCFtoEigenstrat.sh” found here: https://github.com/joanam/scripts/tree/e8c6aa4b919b58d69abba01e7b7e38a892587111
- NOTE: I modified this file to allow non-standard chromosomes names, by indicating a “chom-map.txt” file in the vcftools command
- new file called “convertVCFtoEigenstrat_sd.sh”
- “convertVCFtoEigenstrat.sh” found here: https://github.com/joanam/scripts/tree/e8c6aa4b919b58d69abba01e7b7e38a892587111
- also need in path
- vcftools
- convertf (from eigensoft package)
- conda install -c bioconda eigensoft
- admixtools
- conda install -c bioconda admixtools
Prepared data and run admixtools
mkdir ~/lustre118_link/trichuris_trichiura/05_ANALYSIS/FSTATS
cd ~/lustre118_link/trichuris_trichiura/05_ANALYSIS/FSTATS
ln -s ../../04_VARIANTS/GATK_HC_MERGED/nuclear_samples3x_missing0.8_animalPhonly.recode.vcf.gz
# need to generate a list of scaffold ids, to generate a file called "chrom-map.txt". This is important to make the the scaffold names are parsed properly downstream
bcftools view -H nuclear_samples3x_missing0.8_animalPhonly.recode.vcf.gz | cut -f 1 | uniq | awk '{print $0"\t"$0}' > chrom-map.txt
# run the conversion script.
#--- note have to drop the "vcf.gz" suffix
zcat nuclear_samples3x_missing0.8_animalPhonly.recode.vcf.gz > nuclear_samples3x_missing0.8_animalPhonly.recode.vcf
./convertVCFtoEigenstrat_sd.sh nuclear_samples3x_missing0.8_animalPhonly.recode
# need to manually modify the ".ind" file - the thrid column shows "control" where they should show population IDs
# simply cat the file, copy into a text editor, change it, then move it back
# make a new populations file
> admixtools_pops.txt
# set the outgroup
OUTGROUP=BABOON
# loop throguh the populations to generate the pop file as input to admixtools
for i in BABOON CHN HND UGA ANCIENT; do
for j in BABOON CHN HND UGA ANCIENT; do
if [[ "$i" == "$j" ]] || [[ "$i" == "$OUTGROUP" ]] || [[ "$j" == "$OUTGROUP" ]]; then
:
else
echo -e "${i}\t${j}\t${OUTGROUP}" >> admixtools_pops.txt;
fi;
done;
done
# I manually removed duplicates here
# after looking at the data from the first analysis in R, also decided to set HND as an outgroup.
OUTGROUP=HND
for i in BABOON CHN HND UGA ANCIENT; do
for j in BABOON CHN HND UGA ANCIENT; do
if [[ "$i" == "$j" ]] || [[ "$i" == "$OUTGROUP" ]] || [[ "$j" == "$OUTGROUP" ]]; then
:
else
echo -e "${i}\t${j}\t${OUTGROUP}" >> admixtools_pops.txt;
fi;
done;
done
# run admixtools to generate f3 stats
qp3Pop -p PARAMETER_FILE > qp3Pop.out
# parse the output so it is user friendly to plot
grep "result" qp3Pop.out | awk '{print $2,$3,$4,$5,$6,$7,$8}' OFS="\t" > qp3Pop.clean.out
- where “PARAMETER_FILE”:
genotypename: nuclear_samples3x_missing0.8_animalPhonly.recode.eigenstratgeno (in eigenstrat format)
snpname: nuclear_samples3x_missing0.8_animalPhonly.recode.snp (in eigenstrat format)
indivname: nuclear_samples3x_missing0.8_animalPhonly.recode.ind (in eigenstrat format)
popfilename: admixtools_pops.txt
inbreed: YES
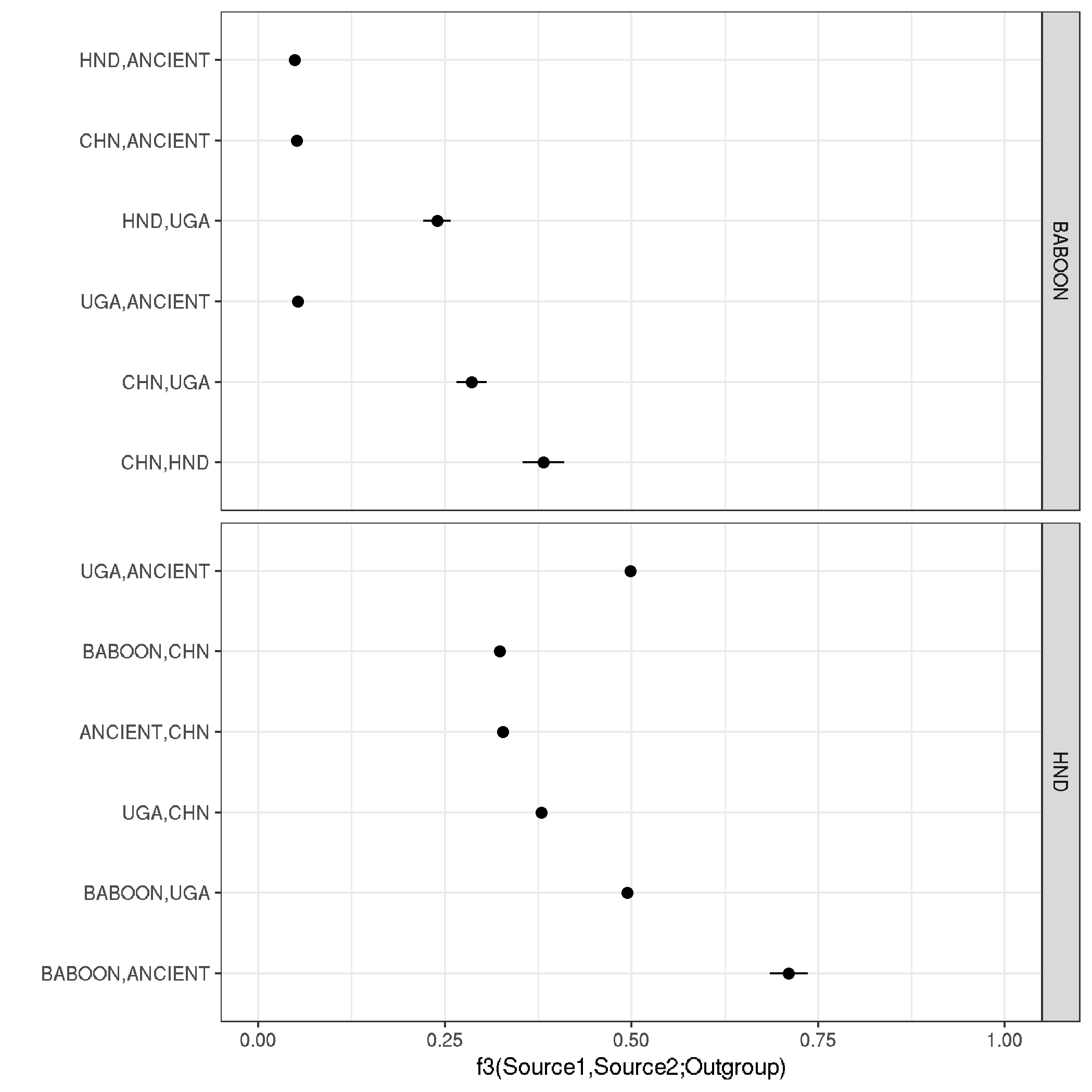
plotting admix data
# load libraries
library(tidyverse)
# read data
data <- read.delim("qp3Pop.clean.out", header=F, sep="\t")
# fix headings
colnames(data) <- c("Source_1", "Source_2", "Outgroup", "f_3", "std_err", "Z_score", "SNPs")
# make a plot
ggplot(data,aes(f_3, reorder(paste0(Source_1,",",Source_2), -f_3))) +
geom_point(size = 2) +
geom_segment(aes(x = f_3-std_err, y = paste0(Source_1,",",Source_2), xend = f_3+std_err, yend = paste0(Source_1,",",Source_2))) +
theme_bw() + xlim(0,1) +
labs(x = "f3(Source1,Source2;Outgroup)" , y = "") +
facet_grid(Outgroup~., scale="free_y", space = "free_y")
# save it
ggsave("plot_admixtools_f3_statistics.png")
ggsave("plot_admixtools_f3_statistics.pdf", height = 4, width = 5, useDingbats = FALSE)